
Using the supercomputer to look for traces of evolution in genes
In the HP2C project “Selectome”, scientists from the University of Lausanne and the SIB Institute of Bioinformatics and members of CSCS have successfully revised a code. It now also runs on supercomputers and calculates selection events in genes many times more quickly.
by Simone Ulmer
A small change in the protein Opsin of the visual pigments once caused primates, including man, to become sensitive to different wavelengths of light and see colours today. The mutation of the gene that encodes Opsin is a simple but typical example of positive selection according to Darwin. The change in the organism brought about a better adaptation to the environmental conditions and thus an advantage. Due to the mutation in the Opsin gene, unlike other mammals primates can see colours today and distinguish between ripe and unripe fruit when foraging for food. As the change proved successful, it was passed on to subsequent generations.
Scanning the Tree of Life
For evolutionary biologists, it is important to find molecular traces of evolution in a genome to gain an insight into the process. Moreover, the events provide key information on how genes work. Marc Robinson-Rechavi from the University of Lausanne and his team are now using supercomputers to look for traces of evolution in the Tree of Life, which symbolises the development of life, in vertebrate genes.
To trace the positive mutations – which mostly took place millions or even several hundreds of millions of years ago – in a gene, the researchers use a particular computer programme that scans the gene’s sequences for possible candidates for a positive mutation with the aid of calculations based on complicated mathematical models of how genes can evolve. The scientists verify which models fit closely with an adaptive modification of a gene. In the calculations, the possibility of different kinds of evolution repeatedly has to be tested on different areas of the protein. If the researchers find a potential candidate, they save the results of their analyses in the Selectome database.

- Species adapt to environmental changes, which is reflected in the appearance of certain body parts. In this case, the pecker of a bird has changed.
Exponential increase in data
Various research groups have been using computers (initially with conventional PCs) to look for traces of positive selection in decoded genes for around ten years. “However, the data set the calculations are based upon is growing exponentially,” says Robinson-Rechavi. The gene database is updated every two and a half months. Depending on how much information or how many species are taken into consideration, the systematic “scan” of families of gene sequences can thus take far longer than three months. However, it is precisely the consideration of a large amount of information that increases the statistical significance of identifying the right molecule where the positive selection took place.
When Robinson-Rechavi took up too much of the Vital IT cluster (bioinformatics competence centre and HPC at the SIB) with his extensive calculation, for Heinz Stockinger, a senior scientist and project manager, it was clear that the software needed to be more efficient. Stockinger and his team thus proposed revising the software codes so that they could run on a supercomputer. The code had to be rewritten in such a way that several calculations could be performed in parallel. As a result, the team enlisted the support of the CSCS through a successful application for an HP2C project, which came in the form of computer scientist Mario Valle. Unlike with classic HPC applications, the amounts of data to be considered might well increase exponentially for the Lausanne scientists and the researchers make purely statistical calculations as opposed to simulations. Nevertheless, the aim and significance of the project managed to win over the sponsors in HP2C.
Today, Robinson-Rechavi, Stockinger, and third co-investigator Nicolas Salamin, seem more than happy since they have succeeded parallelising the code despite the difficult requirements: according to Mario Valle, the Tree of Life has a completely different data structure to around ninety-nine per cent of the projects that are computed at the CSCS. “Normally, the problems are calculated ‘on grids’, but that’s not possible with the Tree of Life,” stresses the computer scientist. They therefore had to subdivide the Tree of Life data in an entirely different way compared to the one used when calculating the weather.
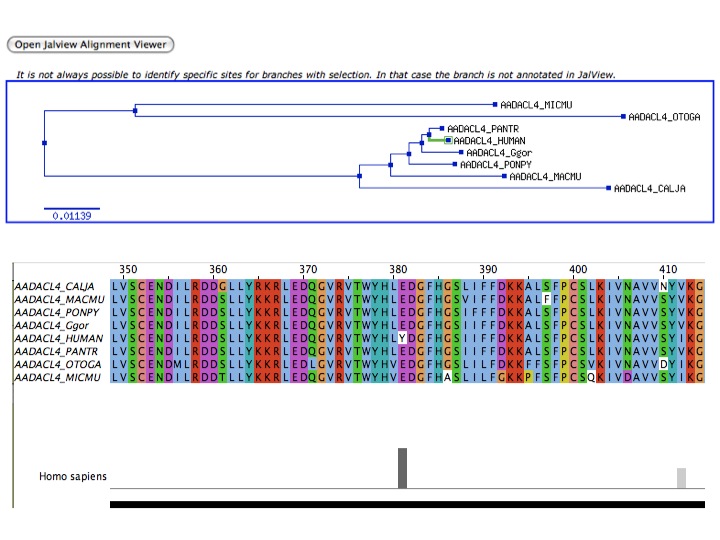
- Details of a positive selection event as displayed on the Selectome website. In the top part, the phylogenetic tree of several species is shown inclusive the positive selection event in green. In the lower part, details on the actual gene sequences are given.
Parallelised code sixty times faster
The first tests of calculations with the new code are now running on the CSCS supercomputer Monte Rosa: the supercomputer scans the data for traces of positive selection sixty times faster than before. During the test runs on 32,257 processor cores – which correspond to a utilisation of way over half the supercomputer – plant genes from 506 species are being scanned. Although there are 407 possible points on the genes where positive selection could have occurred, the calculation only takes about eighteen minutes, Valle explains. One problem – albeit general in evolutionary biology – is that it is not possible to verify the accuracy of the calculations. “We can carry out simulations to demonstrate that, under idealised conditions, our calculations are correct. But we make our simulations based on the same assumptions we use in our models, so it isn’t really a proof,” says Robinson-Rechavi.
The original software comes from University College London and is constantly being refined. “The goal, however, isn’t efficiency, but to develop increasingly elaborate models that illustrate biology very precisely,” says Robinson-Rechavi. Nonetheless, the scientist and his team would solely like to find evidence of positive selection. His aim is to test the models with the new code and scan the roughly 20,000 vertebrate genes for positive selection. The team is hoping for the first results in June.
Even if the study is currently pure basic research that should help to understand the evolution of life and how genes function better, one day the results obtained could yield one of the many puzzle pieces that are useful for health science; of that Robinson-Rechavi and Stockinger are convinced. “The aim today isn’t to prolong life by a couple of years, but rather to enable people to grow old as healthily as possible,” says Robinson-Rechavi. Consequently, it is important to understand the processes in the human organism as accurately as possible, especially at molecular level. The data in the Selectome database is therefore available to all researchers.
| Procedure |
|---|
| For the calculations, the scientists use decoded genes from the so-called Ensembl database. The possible transition from one gene function to a new one is calculated with Markov models based on estimations of parameters. One of these parameters is a quotient of the mutation rate, genetic drift and time to mutation rate, genetic drift, time and selection pressure. These are each determined from the number of changes or abilities in a gene that are the same or not. |